Disclaimer: This article was co-authored by Bruce Kneller and Mike.
On August 12th 2016, the FDA published a new Draft Guidance Document for NDIs (New Dietary Ingredients), in the Federal Register. Since this document is 102 pages long, there is much to discuss.
TL;DR
The FDA's proposal is nothing short of a multi-pronged attack on the entire dietary supplement industry, and reaches much further than the bodybuilding or sports nutrition sectors.
It does not address the core problems that consumers face, which occur due to the FDA's lack of enforcement of existing laws. Instead, the FDA is attempting to add mountains of costly and unnecessary bureaucratic processes that are literally impossible to achieve the remaining good actors of the industry.
-
No Reaction... Because Nobody Read It?
Despite the lack of reaction from industry insiders thus far (likely due to the fact that few people have actually read this monstrous document), there is much to be alarmed over.
This document represents an extreme amount of executive overreach from the federal government, and is purposefully vague yet simultaneously filled with hostile legal traps aimed towards damaging American businesses and limiting consumer choice.
Worse, we do not believe it represents the spirit of DSHEA 1994 as enacted into law some 22 years ago.
-
Ingredients That Could Become Illegal
The following ingredients are just a few that could become illegal for use in dietary supplements if this document is enacted “as is”:
- Molecularly Distilled Fish Oil
- Fish Oils with Unnaturally High EPA:DHA Ratios
- Hydrolyzed Whey Protein
- Ubiquinol
- Ashwagandha
- Resveratrol
- Bacopa monnieri
- Theanine
- Yohimbe
- Synephrine
- Octopamine
- Hordenine, and
- N-Methyltyramine
...amongst potential others, despite each of these ingredients having a true basis in nature and being used in the dietary supplement marketplace with a high degree of safety for decades.
Increasing costs: Other ingredients such as Taurine and Vitamin C from Rose Hips could become monumentally more expensive, and this may subsequently have a profoundly negative effect on neighboring industries such as pet food products.
These ingredients are used by tens of millions of people in the United States each year - with most usage entirely unrelated to sports nutrition and bodybuilding.
-
The Ramifications: Yet Another Industry Sent Overseas
If passed, the American dietary supplement industry could forever be ruined, shifting more economic and labor resources overseas where such bureaucracies do not occur.
Further, the writing of such vague, byzantine laws will likely create unintended consequences that we cannot even begin to imagine today, much of which will be fueled by more intra-industry & class action lawsuits that will only benefit tort lawyers.
-
Get involved and Contact your Representatives in Congress!
As such, we urge our readers to write your representatives in Congress.

As the title page states, this is not yet law and is open for comments through October 11, 2016. We ask you to do your research and write your representatives in Congress!
While it seems that well-respected industry luminaries like Daniel Fabricant from the Natural Products Association and Steve Mister from the Council for Responsible Nutrition seem somewhat or partially satisfied by the content of the new FDA document, we are not. Parties that "approve" of the document were too quick to do so, raising our suspicion that they did not fully understand it nor fully read it before proffering public comments.
Ultimately, this is a terribly-written document, and we will explain most of the problematic issues it contains in piece-by-piece fashion, capturing the overly vague statements, the catch-22's, and the legal gotcha’s that will be a boon only to litigators while potentially ending the American dietary supplement industry as we have known it for decades.
So let’s take a look at some of the more concerning aspects of this new, draft (yes, it is still non-binding) FDA Guidance Document for NDI’s (hereafter FDAGD) that are nothing but “problematic”.
What is a dietary ingredient? We still do not have actual definitions!
Page 13 of the new FDAGD reads upon part of DSHEA 1994 and 201(ff)(1) of the FD&C Act (21 U.S.C. 321(ff)(1)) regarding what is a dietary ingredient. You have almost certainly seen this before:
As defined in section 201(ff)(1) of the FD&C Act (21 U.S.C. 321(ff)(1)), a “dietary ingredient” is any one of the following:
- (A) A vitamin;
- (B) A mineral;
- (C) An herb or other botanical;
- (D) An amino acid;
- (E) A dietary substance for use by man to supplement the diet by increasing the total dietary intake; or
- (F) A concentrate, metabolite, constituent, extract, or combination of any ingredient described in (A), (B), (C), (D), or (E).
An NDI is defined as a dietary ingredient that was not marketed in the U.S. before October 15, 1994 (21 U.S.C. 350b(d)). Thus, to be an NDI, a substance must be a dietary ingredient. [emphasis ours]
Unfortunately, the draft guidance does absolutely nothing to clarify the vagueness that has forever plagued this document. What’s the exact definition of a vitamin?How about Line (E) or Line (F) above? How many steps down the line is something still considered a "metabolite"?
And how do you define the word "combination" in line (F), considering that the FDA has taken action against Picamilon, which is a safe combination of two other permissible compounds (Niacin and GABA)? Does the FDA even follow their our own guidelines, or are they only capable of writing more that they cannot or will not enforce?
We need clarity, not additional mud
Until we can get specific, legal, and understandable definitions for what a dietary ingredient actually is, it is basically an exercise in futility to try to articulate what a dietary ingredient is surely not.

It seems that the FDA is purposefully attempting to legally pigeonhole the industry into absolutely impossible positions. See the Catch-22 Below.
Without solid clarification, we are immediately stuck in the legal mud. The FDA comments starting on Page 13 of the FDAGD reads that NDIs can only be filed for things that meet the above "definition" of what a dietary ingredient is, but you can’t file an NDI for something that is not a dietary ingredient (or potentially a dietary ingredient)!! Yet these ingredients have never even been defined -- they are only categorized, at best!
Confused yet? We are hardly past the introduction and the FDA has already presented us with circular reasoning that is ultimately only open to interpretation from a federal court and anyone with enough legal firepower to get their case heard there.
The FDA's New Pre-DSHEA “Catch-22”
Interestingly, the FDA takes a position on pre-DSHEA 1994 (stuff sold before the “anointed date” of October 15, 1994) moieties, stating that even if the moiety was sold in the USA as a food stuff before the anointed date, if it was not specifically sold as a dietary ingredient in a dietary supplement before that date (meaning if you found the moiety in an apple or a piece of chicken but the moiety itself was not inside some container clearly marked and sold as “dietary ingredient/dietary supplement”) then you still need to file an NDI application for the ingredient (or play the FDA-certifies-as-GRAS kabuki dance through the back door...more on that later!)
Since there was no legally codified definition of what exactly a dietary ingredient or dietary supplement was during the pre-DSHEA 1994 days (remember, DSHEA 1994 was enacted for that very reason), the FDA preemptively comments:
We recognize that the present definitions of “dietary supplement” and “dietary ingredient” were not added to the FD&C Act until after October 15, 1994, and that many products now marketed as dietary ingredients for use in dietary supplements were marketed under other product categories, such as foods for special dietary use or food additives.
Therefore, we interpret “dietary ingredient” to refer to ingredients that (1) if marketed today, would qualify as “dietary ingredients” under 21 U.S.C. 321(ff)(1); and (2) when marketed before October 15, 1994, were intended for use as or in a product that would now be a “dietary supplement” as defined in 21 U.S.C. 321(ff) and that would not also meet the definition of a drug. [emphasis ours]
But once again, we still don't have actual definitions for dietary supplements or dietary ingredients, as noted above (i.e., what exactly is a vitamin?) In essence, the FDA is using undefined terms to define newly undefined terms.
What you're seeing here are two logical fallacies in one: the first known as Begging The Question and the second known as an Etymological Fallacy.
It goes without saying that this is never a good way to clarify an idea or legally binding concept for public consumption. We should never have to deal with "shoulda woulda coulda" situations nor "time machine paradoxes" inside of legal binding documents.
Moving on to the second part of that section, how does one prove that something was marketed before the anointed day? Beginning on page 17 of the FDAGD we see the following:
What documentation does FDA recommend to show that a dietary ingredient was marketed prior to October 15, 1994?
Documentation to show that a dietary ingredient is not an NDI should consist of written business record, promotional materials, or press reports with a contemporaneous date prior to October 15, 1994. Examples include sales records, bills of lading, sales contracts, manufacturing records, commercial invoices, magazine advertisements, mail order catalogs, or sales brochures. Documentation should include adequate information to establish that marketing took place in the U.S.: the identity (e.g., chemical or botanical name) of the marketed in whether the ingredient was marketed as a dietary ingredient or for some other purpose. For example, advertising in bodybuilding magazines could be adequate evidence of marketing as a dietary ingredient. [emphasis ours]
This type of requirement favors certain companies over others in codified form - which is undoubtedly against the spirit of American commerce.
So if a company was not around before the anointed date or did not sell a particular ingredient labeled clearly as a dietary ingredient in a dietary supplement before that date? This will be “problematic” again.
Since the FDA wants each company marketing a dietary ingredient to hold proof of pre-DHSEAness or file an NDI application, we could have a situation where an ancient company has proprietary documents demonstrating that a particular ingredient is exempt by anointed date while a newer company may not ever be able to get FDA to accept an NDI submission for the same exact ingredient and could find itself precluded from marketing it ever.
This type of requirement favors certain companies over others in codified form -- which is undoubtedly against the spirit of American commerce. We even argue that it is illegal due its anti-competitive nature.
Introducing the FDA's "Time Machine Logic"
But let's say you are able to dredge up old copies of Muscle & Fitness or Iron Man and can find an advertisement somewhere in the tattered pages of a magazine that reads on the exact, specific identity of something. Well then you do not need that NDI application. Bravo!But if you can’t do so? Tough cookies – you are filing a NDI application to potentially remain compliant!Assuming what you want to sell is even “NDI eligible” - an additional labyrinthe to traverse.
But who back in 1994 was calling it “Pausinystalia johimbe aqueous extract” when they were not legally required to do so at the time?!
The FDA probably knows full well that this exact, specific terminology they want to see was likely rarely used, if ever, before 1994 (after all, it wasn't necessary from a legal standpoint), and is purposefully using these new "time machine guidelines" to remove whole cloth, several entire classes of ingredients right out of the industry, hoping that nobody will publicly object or contact their congressperson or senator. We have different ideas though...
Anti-Competitive Exclusivity: Sorry, You Don’t Get In – You Aren't On “The List!”
A lot of us deeply involved in the formulation side of the industry rely on an authoritative book published in 1992 called Herbs of Commerce, authored by Moley, et al, to substantiate that indeed, something qualifies as pre-DSHEA.After all, 1992 came before 1994, right?And “herb” and “commerce” would be understood by anyone who has a third grader’s command of the English language as “the marketing and sale of stuff that is derived from plants.”
However, FDA clearly is not having this, as the FDAGD reads:
Although references published before October 15, 1994, such as the 1992 edition of Herbs of Commerce, may be supportive, we are unlikely to regard a listing in Herbs of Commerce as being solely determinative of whether a dietary ingredient was marketed as such before October 15, 1994 because this listing may not specify necessary information such as the plant part and/or extract type. If you rely on Herbs of Commerce as evidence that your dietary ingredient is not an NDI, we recommend that you maintain additional documentation showing that the botanical was marketed as a dietary ingredient before October 15,1994. The documentation should specify the plant part from which the botanical dietary ingredient was derived, and for botanical extracts it should also specify the extract type.
So in essence, using our Yohimbe example from above, in order for this to qualify as pre-DSHEA 1994 exempted, the seller today would ostensibly need to show something like the following in an advertisement or some B2B/B2C business documentssomewhere before the anointed day:
But surely somebody -- somewhere -- has some sort of a list of that reads on a lot of stuff that was sold as dietary ingredients -- specifically as dietary ingredients (whatever the heck that means anymore) - before October 15, 1994 - a list that FDA views as authoritative?
Is there an authoritative list of dietary ingredients that were marketed prior to October 15, 1994 (a so-called “grandfathered list” or “old dietary ingredient list”)?
Not currently. Some trade associations and other industry groups have compiled lists of “old dietary ingredients," though FDA is unable to verify the accuracy of these lists[...]The lists contain ingredients FDA believes are...mixtures that are only vaguely described, like “sterol complete premix.”
[...]FDA does not accept the inclusion of an ingredient on an industry list of pre-DSHEA dietary ingredients as proof that the ingredient is not an NDI. However, in response to comments, we are prepared to develop an authoritative list of pre-DSHEA ingredients, based on independent and verifiable data. Because FDA does not generally have access to marketing records for dietary ingredients and dietary supplements, the documentation of pre-DSHEA marketing would have to be supplied by industry.
FDA’s current thinking is that the two main factors for placing an ingredient on an authoritative list of pre-DSHEA ingredients would be: (1) adequate documentation of marketing for use as or in a dietary supplement in the U.S. before October 15, 1994: and (2) a precise description of the identity of the ingredient marketed. Records offered to support an item’s inclusion on the list should specify the date of marketing in the U.S. and clearly identify the Ingredient marketed on that date. Documentation of an ingredient’s identity should be sufficiently precise to uniquely identify the ingredient.
[...]each dietary supplement manufacturer and distributor is responsible for determining whether each dietary ingredient in each of its dietary supplements is an NDI and ensuring that the firm complies with the NDI notification requirements, if applicable.
Point being? The FDA may be set to stage a multi-pronged attack on the entire dietary supplement industry.
...it appears the FDA has created a purposefully vague document and then added exclusionary exemptions and stipulations to ensure that the vagaries are in favor of their political agenda..
This prong here insures that proving a dietary ingredient is grandfathered because it was marketed or sold pre-DSHEA 1994 is an exercise akin to untying The Gordian Knot.You have to show very exacting, highly detailed documentation that will be at least 22 years old if it even exists anywhere anymore or existed in the first place.
By refusing today to accept any current and encompassing list as an “authoritative” list - and making it public - FDA can then essentially force every company selling creatine monohydrate as a dietary ingredient in a dietary supplement to absolutely prove that it was marketed/sold before October 15, 1994. That may seem to be an extreme example but FDA has been known to do (or “not do”) some pretty extreme things when it comes to the industry.
Read their words in their entirety
But wait, they stated that they are "prepared to develop an authoritative list of pre-DSHEA ingredients"! Hold on and read that closely -- it does not mean they will create such a list - they are merely "prepared" to do so. The FDA's legal eagles have put several such linguistic traps all over this document.
In response, we ask, “Prepared when? Prepared how? Where will it be published? Will it be public? How often will the list be edited or updated if new evidence comes to light?”
If these questions are not answered, then we should only take them at their word - they're "prepared" to make a list but have absolutely no legal obligation to do so - and thus likely won't.
As we've seen in the past, the FDA are experts when it comes to convenient inaction. The more closely you read this document, the more apparent the opportunity for industry failure becomes.
Ultimately, it appears the FDA has created a purposefully vague document and then added exclusionary exemptions and stipulations to ensure that the vagaries are in favor of their political agenda.
The GRAS is not always Greener On The Other Side of The Fence!
-
The first way is someone submits a dossier to the FDA (similar to the NDI submission) and FDA reviews this, agrees the stuff is GRAS when used per the constraints in the submission.FDA then publishes their opinion of GRASness in the Federal Register.
-
The other way, which is usually the “way of choice” for many in the dietary supplement realm, is to establish a panel of “acknowledged experts” – usually semi-independent academics and professors – to supposedly review your stuff and determine through what is essentially a self-published White Paper that in their “expert opinion” that the stuff is indeed, generally regarded as safe in the way someone/you want to sell or market it.
While the expert or “self affirmed” GRAS declaration might raise an eyebrow – and we can and should debate that - surely something the FDA reviews and publishes as “A-OK” in the Federal Register as GRAS is like gospel and can be used as a dietary ingredient, right? Maybe? FDA’s FDAGD reads thusly:
Am I required to submit an NDI notification for a dietary ingredient that is an NDI, but has been (a) listed or affirmed by FDA as generally recognized as safe (GRAS) for direct addition to food or (b) approved as a direct food additive in the U.S.?
No, as long as the following conditions are met. The direct food additive or GRAS substance (1) has been used in the food supply (i.e., in conventional foods) and (2) is to be used as a dietary ingredient without chemical alteration. If the NDI has been legally marketed in the U.S. as an ingredient for use in conventional food and has been introduced into the food supply as a result of such marketing, it would be exempt from the notification. [emphasis ours]
There are numerous people/companies who are selling products they would consider as “completely legal dietary ingredients” because they have either self-affirmed as GRAS or went through considerable trouble to have FDA “bless” their idea/stuff as GRAS in the Federal Register.If the above opinion stands, this will no longer be the case unless the ingredient has been used in the USA as a direct food additive or GRAS substance directly added to a/or as a conventional food without "chemical alteration" first!
So let’s say there is an ingredient naturally found in tea (Camelia sinensis) that someone managed to extract or synthesize (more on that later) and had declared as GRAS, even by the great minds at FDA!Despite the GRAS declaration by FDA, the mere presence of the ingredient in tea is not enough for it to be considered a dietary ingredient. Per the new guidelines, it's perfectly safe for use in conventional food, but all of the sudden that doesn't mean it's safe or legal as a dietary ingredient.
The extracted/synthesized ingredient would now need to be used or added to something else that is used in the conventional food supply in the USA BEFORE it could be used as a dietary ingredient added into a dietary supplement formulation.
Ironically, something can potentially be GRAS if "chemically altered," but now it cannot be submitted on an NDI application -- which brings us to our next section:
Altered States?
What are examples of processes that chemically alter an article of food present in the food supply?
Below are some examples of processes that FDA would likely consider to involve chemical alteration. These processes would also be likely to affect the safety profile of a dietary ingredient. The examples below are intended only for the purposes of illustration and are not a comprehensive list of processes that result in chemical alteration:
- A process that makes or breaks chemical bonds, unless the bonds created by the process are reversed when the ingredient is dissolved in water (e.g., creation of a soluble salt) or during ingestion. Example: hydrolysis.
So simple ionic salts may be allowable.Finally, something positive from the FDAGD! Continued:
- Removal of some components of a tincture or solution in water, which changes the chemical or molecular composition or structure of the mixture. Examples: chromatography, distillation, and filtration.
So is the FDA going to hold the opinion that some cold & micro-filtered whey protein concentrates or isolates are never acceptable as a dietary ingredient and can not be submitted as an NDI? Note - this does not mean they can not be found in or as conventional foods, it just means they can not be dietary ingredients in dietary supplements via the NDI application route. They would have to be approved as FDA beknighted GRAS substances first, used in or as a conventional food in the USA next and then they would be OK - maybe - as dietary ingredients.
...With each new paragraph, more questions are introduced - not answers. This is great business for bureaucrats and lawyers, but it’s a total market destroyer for everyone else...
This seems 1) confusing and 2) asinine. This is however, very likely creating a loophole FDA did not anticipate when they authored the FDAGD.
There are a plethora of new technologies (e.g., post anointed day) that are used to separate out various whey protein fractions that obviously alter the molecular composition of the protein and amino acid content advantageously. If this document stands “as is” it is possible the FDA could take the position that they are actually adulterated and/or drugs!
It gets worse, too: what about acid hydrolyzed whey proteins? The use of an acid, which is neither water, ethanol or a simple filter, absolutely and undeniably alters (“chemically alters”) the composition of whey protein!
- Use of solvents other than water or aqueous ethanol to make an extract or tincture. The official legislative history of DSHEA specifies that “solution in water” and “tincture” (solution in aqueous ethanol) are not processes that chemically alter a food. However, other solvents typically alter the composition of the extract in significantly different ways, usually by extracting different types of constituents than are extracted using water and aqueous ethanol.
- High temperature baking or cooking of an ingredient that has not previously been baked or cooked, unless the process causes only minor loss of volatile components with no other changes to the chemical or molecular composition or structure of the ingredient.
So at what temperature does the FDA consider something “high temperature?” How minor is something allowed to be in order for FDA to consider it “minor loss?” If I have a wet extract or concentrate of an herb, does this mean I can’t use a heat- drying machine or rotary evaporator to evaporate off excess water or solvent? Only natural, “air dry” techniques are OK? What if the day I dry my stuff is a really dry and hot day?Maybe we’ll see “drying” facilities pop up all over Southern Texas, Arizona and New Mexico?
Just like every other overly voluminous law written lately, it seems that with each new paragraph, more questions are introduced -- not answers. This is great business for bureaucrats and lawyers, but it's a total market destroyer for everyone else - especially well-meaning small businesses.
Bad Interpretations: Watch the FDA's new guidelines eat itself
Consider the following opinion as written into the FDAGD:
In general, FDA considers a process that does not result in chemical alteration to mean a process that: (1) involves an ingredient composed of one single raw material, or derived from a single raw material using a manufacturing process that involves only physical steps (e.g., water extraction and condensation); and (2) does not involve attempts to selectively increase the concentration of particular active ingredients...
So if you wanted to say, increase the concentration of a certain flavone or terpenoid in an herbal extract from say, 0.005% w/w (mass fraction) to something like 5% or 10% w/w, even if you could use only a simple water extraction to do it, the FDA considers the result "chemically altered".
And if it's chemically altered, even using this officially-approved law-abiding method, it still can’t be a dietary ingredient, so you can’t submit an NDI with the hopes of making it one. Welcome to our next catch-22 - this guideline literally eats itself!
Although maybe, you could submit to FDA as a GRAS substance, get them to agree, have them publish in the Federal Register, use the substance in or as a conventional food first and THEN you can market as a dietary ingredient.How many hoops did we just jump through?
As you can see, every which way we turn, ingredients that have been known to be both safe and effective in the promotion of health are now possibly in question and at risk for administrative enforcement by FDA. And if not enforced by the FDA, then surely the class action lawyers who feed on such vagaries will prepare to feast - but more on that later.
Next, let's dive deeper into some examples to see just how bad this could get.
Part II: Getting more specific
Thus far, we've discussed some aspects regarding the New FDA Draft Guidance Document for NDI’s (FDAGD) that we feel are genuinely problematic – either they are vague, baffling, and/or confusing, or they are so cumbersome that there will be a significant impact on the industry's ability to operate and innovate in the health marketplace.
We have opined on the lack of clarity as to what really would be considered a pre-DSHEA, grandfathered compound that FDA would consider a legitimate dietary ingredient, as well as FDA’s position on what “chemically altered” (and thus, unacceptable for submission as a NDI) may mean. And once again, we've still never been provided with definitions of what these classes of ingredients actually are, making this entire exercise a futile effort right from the start.
But the situation gets even uglier for consumers, industry professionals, and anyone who is not a lawyer.
We want to now discuss even more troubling and restrictive sections of the FDAGD. These sections relate to synthetic versions of moieties that rationally should be acceptable (but as we will soon learn, clearly are not per the FDA’s stance) as dietary ingredients or at least, potential NDIs.
Think That Vitamin C Came From Freshly-Squeezed Oranges?
As some background, there are multitudes of moieties currently marketed/sold as dietary ingredients that are found in nature, but are not typically derived directly from something naturally for commercial sale. That is to say, these compounds are chemically synthesized in a factory or laboratory somewhere.

A caffeine molecule is a caffeine molecule. So how can one be legal and the other be illegal due to the extract process used?
For example, Vitamin C (ascorbic acid) is found naturally in plants (e.g., Rose Hips – see here), but it’s exceedingly expensive to extract Vitamin C from a plant. So instead, less expensive, synthetic manufacturing methods have been developed to enable this ingredient to be sold to the consumer at a reasonable price. You didn't think your Vitamin C came from someone squeezing a Florida orange, did you?
The synthetic version is the same exact molecule, indistinguishable from the natural version. However, the synthetic version comes at a far lower price than the natural version for the consumer. Further, we surmise that it's likely safer due to absolute purity from the manufacturing process, ease of reproducibility, and lack of contaminants. A controlled chemical reaction always behaves in the same manner, but Mother Nature's fruits do not always yield the exact same constituents qualitatively or quantitatively.
Synephrine, octopamine, hordenine and N-methyltyramine are putative neurostimulantsand “fat burners” that are clearly found naturally in the plant, “Bitter Orange” (Citrus aurantium – see here) and have been safely sold for years (if not decades) as “acceptable dietary ingredients” within the industry without any negative enforcement action from FDA until very recently in the case of -maybe- synephrine (see here).
We surmise that most if not virtually all of the synephrine, octopamine, hordenine, and N-methyltyramine we see in the industry are synthetically manufactured and are clearly not materials that have been extracted or concentrated from Bitter Orange. Most are found in dietary supplements as ionic salts, typically as the hydrochloride salt (which would be allowable per FDA as these are typically hydro-soluble in water) because in their natural base forms, they are usually foul tasting, viscous liquids, and/or the base forms have poor shelf life, stability, or other innate characteristics that would effectively compromise their marketability within the industry. Great ingredients found in nature, yet best used when purity can be repeatably controlled and tested. Not to mention the relative and absolute affordability they offer the consumer at large.
Rising costs for the same ingredient
Taurine is also a common ingredient found in both sports nutrition products and other dietary supplements that has been sold as a dietary ingredient without enforcement action by FDA for years if not decades. Taurine is often considered an amino acid but from a technical standpoint (and not the FDAGD or even the United States Code, I might add), it is not really a true amino acid – see here).
Taurine is, however, a naturally occurring substance, being found in mammals/humans/and some plants like sea algae. It's a by-product of methionine and cysteine metabolism. However, we're not aware of any taurine currently being manufactured from cysteine or methionine that's sold in the dietary supplement marketplace or the conventional foodstuff (or even pet food) marketplace, due to the exorbitant cost of such a process.
Instead, most commercial taurine is inexpensively synthesized by the metric ton from 2-hydroxyethanesulfonic acid, that is first synthesized from a reaction of ethylene oxide and sodium bisulfite (none of these are dietary ingredients or food stuffs). Alternatively, commercial taurine is sometimes synthesized from a reaction of aziridine and sulfurous acid. Because of such processes, taurine is quite inexpensive, perhaps $3.00US/kg here in the United States on the wholesale market.
So Are Synthetic Ingredients With Natural Counterparts Acceptable Dietary Ingredients?
One of the burning questions that folks in the industry have been waiting for the FDA to answer has been, “Are synthetic ingredients that are chemically indistinguishable from their naturally found counterparts acceptable as dietary ingredients or potential NDI’s?” FDA’s response to this vexing question is curiously bifurcated and may be a serious source of consternation to the industry for years to come. In the FDADG we find thus:
Under what circumstances does FDA consider synthetically produced substances to be dietary ingredients under the FD&C Act?
Whether a synthetically produced substance qualifies as a dietary ingredient depends on whether the substance fits into one of the categories of dietary ingredients that are defined in section 201(ff)(1) of the FD&C Act (21 U.S.C. 321(ff)(1)).
We need to stop here for a moment because we need to go to that section of 21 USC 321 to look at the categories (and definitions) of dietary ingredients this part of the FDADG refers to.
Look here: https://ods.od.nih.gov/About/DSHEA_Wording.aspx#sec3
Once again we see the mere categorization that masquerades as definition, and still does not truly provide one (a definition):
§3. Definitions.
- (a) Definition of Certain Foods as Dietary Supplements. Section 201 (21 U.S.C. 321) is amended by adding at the end the following:
"(ff) The term "dietary supplement" -
- "(1) means a product (other than tobacco) intended to supplement the diet that bears or contains one or more of the following dietary ingredients:
- "(A) a vitamin;
- "(B) a mineral;
- "(C) an herb or other botanical;
- "(D) an amino acid;
- "(E) a dietary substance for use by man to supplement the diet by increasing the total dietary intake; or
- "(F) a concentrate, metabolite, constituent, extract, or combination of any ingredient described in clause (A), (B), (C), (D), or (E);
And once again, we cannot find any instance in the law that describes what a vitamin or a mineral or an amino acid really is or is not. Nor can we even locate an example of what any of these things are in a non-limiting table or chart.This omission has been problematic for over 20 years, and it continues to be more so each year.
Next in the FDAGD, we see this:
In some cases, description of the category in the FD&C Act encompasses synthetically produced substances; in others, it does not. The six dietary ingredient categories are discussed in the bullets below.
Vitamins, Minerals, and Amino Acids (21 U.S.C. 321(ff)(1)(A), (B), (D))
Synthetic vitamins, minerals, and amino acids qualify as dietary ingredients because vitamins, minerals, and amino acids, regardless of source, are specifically designated as dietary ingredients under sections 201(ff)(1)(A), 201(ff)(1)(B), and 201(ff)(1)(D) of the FD&C Act, respectively. Synthetic vitamins, minerals, and amino acids are recognized as dietary ingredients because a vitamin, mineral, or amino acid is defined by its nutritional function (its ability to provide nutrients to the human body), not by its state of matter like a botanical.
This is where the “curious bifurcation” we alluded to before begins. While we are grateful that FDA believes synthetic vitamins, mineral and amino acids are acceptable for use as dietary ingredients (whatever their respective definitions are or are not), we genuinely do not believe that anyone in authority at FDA could look at us with a straight face and tell us that these three “categories” are defined by “nutritional function” alone – Sodium Chloride, Potassium Chloride and plenty of other mineral salts are indeed, without a doubt, often sold as prescription or over-the-counter drugs which clearly are not defined by their “nutritional function.”
In practically every industry, America is losing control of international markets - and obscene regulatory guidelines like these are exactly why.
These things can and are defined by their pharmacological effects in treating and preventing diseases. The same is true for amino acids and a good many vitamins and their derivatives. Magnesium sulfate or magnesium citrate may be nice way to increase magnesium levels in people in a “nutritional function” sense but magnesium sulfate is also used orally to treat constipation as is magnesium citrate (here). The same is true for many calcium and zinc salts.
Most geologists would consider gold, silver, & platinum to be minerals – we don’t know what the FDA thinks since again, there is no federally codified list of or definitions for “minerals” that we are aware of. We’re pretty sure folks are not consuming these “minerals” or their salts for their “nutritional function” although they do have great value in treating some cancers and autoimmune diseases as prescription drugs. So does this mean we could market and sell a totally synthetic salt of platinum as a dietary supplement so long as we make sure the particular salt we’re selling has never been used specifically as a drug?
Also, it would be helpful if we could see specifically where in the law it reads again that a vitamin or a mineral or an amino acid is defined by its “nutritional function.”
Moving on, here is where the FDAGD springs the proverbial big old “gotcha” on the dietary supplement industry --
Dietary substance for use by man to supplement the diet by increasing the total dietary intake (21 U.S.C. 321(ff)(1)(E)).
For purposes of section 201(ff)(1)(E) of the FD&C Act, we interpret “dietary substance” in accordance with its common, usual meaning [ed. note – Which is? Would have been nice if this was defined somewhere in the law!] because the term is not defined in the FD&C Act or by regulation [ed. note – not defined in the FD&C! No “legal definition!”]. According to Webster’s II New Riverside University Dictionary (1994) [ed. note – since when is Webster’s Dictionary, which is subject to yearly or periodic revisions, legally binding?!], “dietary” means “of or relating to diet” and “diet” means “an organism’s usual food and drink.” In conjunction with “for use by man” we interpret “dietary substance,” as used in section 201(ff)(1)(E), to mean a substance commonly used as human food or drink. The rest of the definition, which specifies that the substance be for use “to supplement the diet by increasing the total dietary intake” is further evidence that “dietary substance” is intended to mean foods and food components that humans eat as part of their usual diet [ed. note – that is one interpretation but not the sole or only plausible interpretation]. One cannot increase the “total dietary intake” of something that is not part of the human diet in the first place.Because the “dietary substance” category is defined in part by history of use, a synthetic copy of a botanical ingredient may qualify as a dietary ingredient under section 201(ff)(1)(E) if the synthetic copy has been used as a lawfully marketed ingredient in the conventional food supply. For example, a synthetic copy of a botanical ingredient would be a dietary ingredient under section 201(ff)(1)(E) if the synthetic copy has been used as an ingredient in the conventional food supply. Two common examples are vanillin and cinnamic acid, botanical constituents that, for economic reasons, are usually produced synthetically for use as flavorings in food.
So, as long as it is present in the conventional food supply and “commonly” eaten by people in the USA in its natural state then a synthetic version might be allowable? What about our previous examples of synephrine, octopamine, hordenine, N-methyltyramine (let’s add in the ever popular yohimbine too)? People eat Citrus aurantium and perhaps, people have consumed the bark from Yohimbe in the form of various teas. How does the FDAGD read upon such?
Concentrate, metabolite, constituent, extract, or combination of another dietary ingredient described in clause (A), (B), (C), (D), or (E) (21 U.S.C. 321(ff)(1)(F)).
A “constituent” is an article that is a physical part of a whole and can be isolated from the whole. A synthetic copy of a constituent of a botanical was never part of the botanical. Therefore, the synthetic copy is not a “constituent” of the botanical and does not qualify as a dietary ingredient under section 201(ff)(1)(F) of the FD&C Act (21 U.S.C. 321(ff)(1)(F)), even if the synthetic copy is chemically identical to a constituent of a plant.
By the same principle, an extract made from a synthetic copy of one or more constituents of a botanical is not an “extract” of the botanical under section 201(ff)(1)(F) of the FD&C Act because the constituents were never part of the botanical and therefore could not be extracted from the botanical. Similarly, a synthetic copy of a botanical concentrate is not a concentrate of a botanical because, by definition, a “concentrate” is an article that has been reduced in volume or bulk by removal of liquid. To make a concentrate of a botanical, one must start by extracting the desired constituents from the botanical with a solvent and then concentrate the constituents by reducing the amount of solvent (e.g., by boiling the extract). If synthetic material that was never actually in the botanical is used as the starting point for a concentrate, the final product will be a concentrate of the synthetic material, not a concentrate of the botanical. For more than a decade, FDA has consistently interpreted section 201(ff)(1)(F) of the FD&C Act as not including synthetic copies of botanical constituents, extracts, or concentrates.
So, first FDA claims that for “more than a decade, the FDA has consistently interpreted section 201(ff)(1)(F) of the FD&C Act as not including synthetic copies of botanical constituents, extracts, or concentrates.”So if this is true, and they genuinely believed this, they rarely, if ever, acted on this philosophy in enforcement actions.We find it a bit fantastic for FDA to claim this.
Executive Overreach?
This also places the FDA, which consist basically of career professionals employed in an administrative agency that is part of the executive branch as“kinda/sorta” acting in a manner that is probably more judicial.We believe firmly that FDA should be enforcing laws – preferably well-written and easy to understand laws - as part of the executive branch and not trying to acting as “first line jurist” by trying to interpret what they believe Congress “really meant 22 years ago” when it enacted legislation that may have been vague.
If a decision should be made and the law is as apparent as it should be or there is a “special circumstance”, then we feel judgment should probably fall under the auspices of the judicial branch (e.g. a federal magistrate/judge/court).
Making NDI Applications Impossible To Even Begin
Next, if the compound (singular) is a synthetic copy of something found naturally in a botanical or herbal product then not only is it likely not a dietary ingredient now (unless it’s pre-DSHEA 1994 or you get them to GRAS-bless it and you put it into a conventional food you sell before trying to market as a dietary ingredient) but apparently, it is completely ineligible for direct NDI application status.
In other words, all synthetic synephrine, octopamine, hordenine, N-methyltyramine, and yohimbine -- not to mention all the other PEA and mild neurostimulants we have come to know and love in our diet pills and pre-workout powders -- will never be afforded the chance to be legal dietary ingredients via an NDI application, irrespective of how many studies demonstrate them to be quite safe, as these are per FDA’s perplexing re-interpretation of DSHEA 1994, ineligible for NDI application.Multitudes of flavones, isoflavones, alkaloids, terpenoids, stilbenoids, and believe it or not, perhaps even agmatine and taurine are at risk here if they are produced synthetically.
Painted Into A Corner
Well, if the synthetic versions are unacceptable then why not just extract them from plants or animals?
As already discussed, this will also not be possible due to the new "chemical alteration" rules! Your extraction/concentration is limited merely to water and ethanol as solvents under the FDAGD.
Also by “selectively trying to increase the concentration of particular active ingredients” FDA is of the opinion you are likely “chemically altering” something and this also disqualifies that something as a dietary ingredient (unless pre-DSHEA/GRAS Blessed and already sold in a food) and prevents it from ever being submitted for consideration as a NDI.
If this stands, the FDADG as written, we are likely to forever lose any truly effective stimulants/fat burning products other than caffeine and its salts as dietary ingredients!
We may be able to save taurine (and agmatine) at great financial cost because:
A metabolite that has been synthesized from another dietary ingredient would be a dietary ingredient under section 201(ff)(1)(F) and could be used as a dietary ingredient in a dietary supplement. Although the definition of a metabolite requires human ingestion of the dietary ingredient to increase the production or flux of the metabolite in the human body, it does not require the metabolism to actually take place in a human being during the manufacture of a dietary ingredient. A metabolite may be synthetically produced, provided that the starting material is a dietary ingredient and the production process mimics the metabolic process in the body following ingestion.
Just so we all fully understand what this means to everyone economically, synthetic taurine costs about $3.00US/kg. To make taurine as a metabolite of l-methionine (which costs $8.00US/kg) or l-cysteine (which costs $18.00US/kg) means you’re looking at taurine costing between $24.00US-$54.00US/kg and the availability of such to be fairly constrained and limited by commercial supplies of l-methionine and l-cysteine.
The Negative Effects Are Worse Than Anyone Seems To Realize
The implementation of this FDADG as it reads now will likely decimate the dietary supplement market if enforced.American companies will lose significant degrees of freedom to operate. Consumer choice will be reduced or fully eliminated in many categories.The economic costs will be manifest as brands/label holders, contract manufacturers, and raw material providers are downsized or shut out of the market altogether. Many ancillary industries such as plastics, paper goods, and transportation will see constriction related to such. Employment and tax revenue could well be reduced.
So why is nobody in the industry talking about it?
We're not sure, but we're not taking this as lightly as everyone else. This is worse than anyone realizes - and that includes those outside of the sports nutrition industry too. Perhaps they haven't taken the time actually read this document... but we have. And it's appalling once you realize how many legitimately beneficial ingredients will likely become illegal if this is passed.
To show that this is going to affect more than just sports nutrition, we've developed a list of "non-bodybuilding" ingredients that will be illegal if this document is brought to life:
Some Ingredients That May Be Illegal/Problematic Include...
-
Molecularly Distilled Fish Oils
Reason: Under Section B, Paragraph 4 of the FDADG, the FDA considers and specifically notes that “distillation” is a process that chemically alters something in a manner that is unacceptable for NDIs.

One of the best fish oils on the market, this one has been "up-formulated" to contain more DHA and EPA - far more effective per volumes of research. And soon to be illegal if this draft is approved.
Who's affected: Nearly everyone, but especially those with cardiovascular conditions and diets that require high amounts of additional omega-3 fatty acids.
Potential Remedy: Would need to be submitted for GRAS approval to FDA, then market & sell as part of a conventional foodstuff before use in dietary supplement formulations and use would be limited to use under constraints of FDA GRAS Approval.
Notes: Unilever has a submitted a GRAS application for molecularly distilled fish oils in 2003. However, to the best of our knowledge it has not been sold in a conventional food as required per the FDADG.
Additionally, currently there is limit of 11.4% maximum distilled fish oils in any food product. Thus, FDA could consider any molecularly distilled fish oil dietary supplement with a concentration exceeding this to be adulterated under section 402(f)(1)(B) of the FD&C Act (21 U.S.C. 342(f)(1)(B))
-
Fish Oils With Unnaturally High EPA:DHA Ratios
Reason: Under Section 5 of the FDADG, “In general, FDA considers a process that does not result in chemical alteration to mean a process that: (1) involves an ingredient composed of one single raw material, or derived from a single raw material using a manufacturing process that involves only physical steps (e.g., water extraction and condensation); and (2) does not involve attempts to selectively increase the concentration of particular active ingredients[...].
Who's affected: Same as above - nearly everyone, especially those with cardiovascular concerns.
Potential Remedy: Would need to be submitted for GRAS approval to FDA, marketed & sold as part of a conventional foodstuff before use as a NDI and would be limited to use under constraints of FDA GRAS Approval.
-
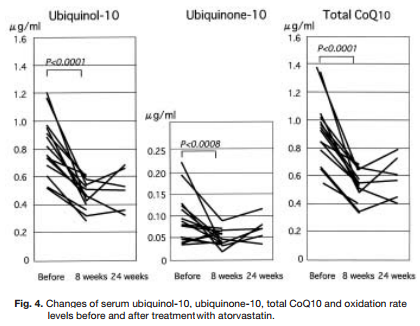
Ubiquinol
Reason: Ubiquinol is a naturally occurring substance that is related to Coenzyme Q10 (CoQ10). The FDA has previously rejected NDI submissions for Ubiquinol citing “[...]the information in your submission does not provide an adequate basis to conclude that the dietary supplement product “ubiquinol” when used under the conditions recommended or suggested in the labeling of your product, will reasonably be expected to be safe.”
While there are companies that have self-affirmed ubiquinol as GRAS, we are unaware of the FDA affirming this substance as GRAS. Even if viewed as GRAS, under the FDAGD, ubiquinol would need to be added to a conventional food stuff that is marketed and sold in the USA before it could be considered an NDI via that avenue.Further, the rejected NDI submission reads on ubiquinol being sold in 50mg oral dose forms (we are unable to see the self-affirmed GRAS submission but our assumption – which may be incorrect – is that that 50mg per serving/dose is what is self affirmed). Thus, even if FDA were to agree with the self-affirmed GRAS status of ubiquinol at 50mg per serving/dose, it could ostensibly be viewed as “adulterated” if sold in 100mg or 200mg per serving/dose forms under section 402(f)(1)(B) of the FD&C Act (21 U.S.C. 342(f)(1)(B)).

Statin drugs greatly reduce levels of ubiquinol, ubiquinone-10, and CoQ10. Ubiquinone supplements can help combat this. So why is the FDA passing a law that will make it illegal and endanger so many prescription drug users?
Who's affected: Statin drug users with high cholesterol. We believe that tens of thousands (if not more) people in the United States use ubiquinol as a dietary supplement to mitigate the effects of some cholesterol lowering medicines (statin-drugs) on mitochondria). While it would instantly make ubiquinol a “drug” if there were any label claims related to it’s use and preventing or mitigating statin-induced side effects (remember, dietary supplements do not prevent or cure any diseases kids per the FDA warning box required on the label of all dietary supplements), this is likely the most common "off-label use" for the product.
Additionally, there is robust research demonstrating numerous health preserving effects of ubiquinol (doses typically range from 90-200mg in such studies).
Potential Remedy: Resubmission and approval of NDI application with more/better data or acceptable GRAS submission to FDA with subsequent use of ubiquinol in a conventional food before NDI use. Particular care should be paid to intended dose/serving size of ubiquinol.
-
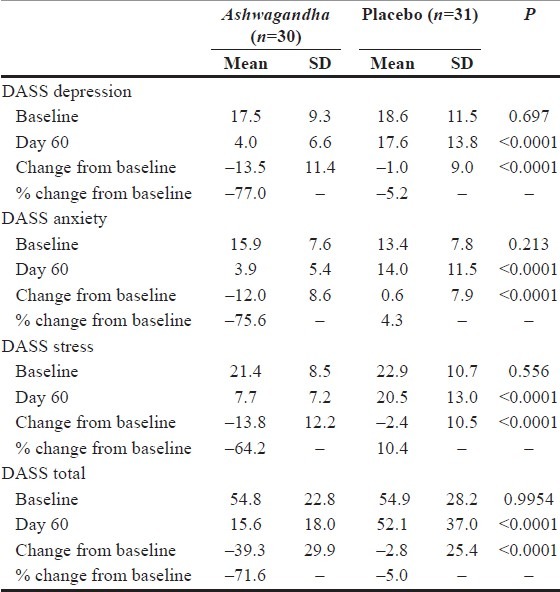
Ashwagandha (Withania somnifera)
Reason: Ashwagandha (Withania somnifera) which is sometimes known as Indian Ginseng is a plant that has been awarded FDA GRAS status for some of its root and leaf extract(s). And while the berries are clearly part of the conventional food supply being used in cheese making, we are unaware of any leaf or root extracts being used in conventional food stuff in the USA.

Ashwagandha can truly reduce levels of depression, stress, and anxiety. Why would the FDA want to make such a great ingredient illegal?? Hmmm....
Who's affected: Multitudes of consumers ranging from those requiring anxiety / stress / antidepressive relief to high-cholesterol patients to those with hormonal imbalances to infertile men.
Potential Remedy: The parts of the plant we want to use as dietary ingredients would need to be marketed & sold as part of a conventional foods stuff before use as a NDI and would be limited to use under constraints of FDA GRAS Approval.
-
Bacopa monnieri
Reason: According to the American Botanical Council in 2011, Bacopa monnieri has NOT been recognized as GRAS by the FDA but they believe “it is acceptable as a dietary supplement component under provisions of the Dietary Supplement Health and Education Act of 1994” citing a book published in 2005 (11 years after DSHEA 1994 was enacted) as possibly being evidentiary. Possibly.
Given the high bar that the FDAGD reads on regarding evidence of pre-DSHEA exemption for botanicals (plant, plant part, how extracted/concentrated, what extracted/concentrate for as well as tangible proof of the marketing of such in the USA before October 15, 1994) we are not yet convinced when push comes to shove that FDA will consider Bacopa as a pre-DSHEA exempt ingredient.
Who's affected: Consumers fighting memory loss and/or cognitive decline and those requiring anxiety reduction or anti-depressive effects.
Potential Remedy: Approved NDI application with FDA or approved GRAS submission and evidence of use in a conventional foodstuff in the USA before use as an NDI.
-
Resveratrol
Reason(s): Resveratrol (trans-resveratrol) is a naturally occurring element found in grapes, blueberries, raspberries, and other plant biomass. An NDI application was indeed submitted to FDA for resveratrol (also see Section 8.0 of this document).
FDA’s official position in regards to that submission was, “[...] your submission does not provide an adequate basis to conclude that resveratrol, when used under the conditions recommended or suggested in the labeling of your product, will reasonably be expected to be safe...”.Further, the FDA is of the opinion that trans-resveratrol is ineligible to be a dietary ingredient anyhow as it is being evaluated (or was) as an investigational new drug (see also Table 3 on page 223 here).
Ironically, there was a GRAS submission to FDA regarding trans-resveratrol after the failed NDI application and FDA generally denying trans-resveratrol eligible to be an NDI although the submitors requested the FDA cease evaluation so it is at least possible for this ingredient to potentially added to a conventional food stuff but likely never as a dietary ingredient in a dietary supplement..
Potential Remedy: None. Our opinion is that trans-resveratrol is not a dietary ingredient and cannot be considered for submission as an NDI.
-
Theanine
Reason: Theanine is a constituent of green tea as well as other plant sources.The FDA has confirmed numerous GRAS submissions for the “L” isomer of theanine as acceptable (synthetically or naturally sourced). The “D” isomer and the racemic mix has not – to our knowledge - been confirmed as GRAS by FDA.
While L-Theanine has been added to 50+ foodstuffs sold in Japan, we are not aware of synthetic L-Theanine or high percentage pure L-Theanine extracts that have been added into the conventional food supply in the USA to date.
Who's affected: This one is a matter of cost savings for consumers. "Theanine" is much cheaper to make than L-Theanine.
Potential Remedy: L-Theanine would need to be added to a conventional food marketed and sold in the USA before it could be considered for use as an NDI.

But the FDA Would Never Take Those Things Away....
One could argue that the FDA would "never take away dietary ingredients” that are so popular with people who use these as adjuncts to mitigate serious conditions such as those using statin-drugs to treat high cholesterol. As we've seen in the past, the FDA's enforcement of its the law is completely unpredictable - if not downright capricious - when it comes to the dietary supplement industry.
Moreover, that argument is now completely irrelevant, because once it's illegal, it's illegal. And if you're breaking the law, even if the FDA doesn't go after you, another batch of friendly faces will: THE TORT LAWYERS.
In Comes The Tort
...This will affect millions of American consumers, and with that it will send more jobs and businesses over the border as well, because we literally can no longer compete legally.
Whether through class action lawsuits or competitors filing unfair competition claims, we expect to see a serious uptick in the amount of civil litigation at the federal level with the ostensible and noble purpose of “protecting the consumer” or “leveling the playing field.”
In late 2014, we predicted that the POM Wonderful vs. Coca Cola Supreme Court decision would lead to several intra-industry lawsuits under the Lanham Act, and we were 100% correct. Sometimes for better, often for worse, those suits have come by and large.
This FDADG gives the tort lawyers a whole new batch of "illegal activities being carried out by good companies with lots of money they can grab at." We expect to see civil lawsuits galore.
A large company might decide it’s a legit business strategy to sue a smaller company to make them “prove” that their product meets all of the new conditions the FDADG imposes on the industry. So, you better be able to substantiate that creatine is pre-DHSEA 1994 exempt or you better be able to show it’s GRAS or you filed an NDI for it or...
Unintended Consequences of Burdensome Bureaucracy
These are just some of the unintended consequences that we have seen, time and time again, by cumbersome laws and executive overreach. Good and safe ingredients will either become illegal, extraordinarily expensive, or worse -- they'll only be available via gray/black markets from overseas sources. So not only will we be shipping more jobs overseas - consumers will be sending more of their hard-earned money there too. A total net-negative for America.
American consumers and industry professionals alike need to realize that this is not just an attack on sports nutrition and bodybuilding supplements -- it will affect millions of American consumers, and with that it will send more jobs and businesses over the border as well, because we literally can no longer compete legally.
In practically every industry, America is losing control of international markets - and obscene regulatory guidelines like these are exactly why. It needs to stop, and it needs to stop right now.
So Now What? Our Calls to Action
Make your voice heard:
If you take issue with any of the items on this page, then Contact the FDA and Congress about the New NDIs.
Commentary was only open until October 11, 2016.
Addendum: "If We Can't Have It, Neither Can They!"
In 2014 in the USA, SCOTUS in an almost unanimous ruling (Justice Scalia, may he rest in peace, was the only dissenter) heard a case involving Myriad Genetics, Inc., in their attempt to patent certain DNA sequences.As perhaps an unintended consequence of the ruling on that case, the US Patent and Trademark Office issued a guidance document for implementation.

GHB (Gamma-Hydroxybutyric acid) was banned in 1991 after a huge media attack as the "date rape drug"... and then mysteriously becomes a prescription drug for narcolepsy in 2002. What else is going to 'disappear' and then' reappear' as a prescription?
It basically reads that if you discover “something” that was not known in any matter before but it is part of nature (meaning you did not synthesize it and it’s not essentially “new” in the universe – it is “naturally occurring”) then it likely can not be patented because any properties that this “something” you discovered possess are naturally-occurring and inherent in the “something”, and while you "discovered" it, you did not "invent" it (there are subtle but important legal and semantical differences).
This ruling had widespread and dramatic impacts for Big Pharma because now, anything Big Pharma discovers that is naturally occurring really can’t easily be patented and developed as a drug or diagnostic for sale. A “tweaked” version of “something” that is somehow – chemically, physically – different from the naturally occurring thing can potentially be patented but the naturally-occurring “something” likely can not.
Consider this: If Big Pharma can not patent a “something” (as in an “extract” or “concentrate”) found in a plant or an herb or an animal, even if the “something” was never known before and just discovered today, Big Pharma is not likely to pursue developing that “something” into a drug or diagnostic. Big Pharma likes and enjoys their monopolies. They virtually never spend money developing “something” they do not have iron-clad patents claims for and this, absolute control over.
Note: In the USA, Big Pharma needs FDA approval to market and sell any drug.
Fast forward two years – it’s 2016 and the FDA (the sole approver of Big Pharma’s patent protected “somethings” in America as salable drugs) issues a new draft guidance document for dietary ingredients that reads that “somethings” from botanicals/herbs can not be synthetic copies of one or two chemicals found in the botanical/herb. Further, we can not extract/concentrate the botanical/herb as to dramatically raise the level of a “something” and have it be an accepted, dietary ingredient.
So? We’ll end this with a quote from a character of a Japanese Manga cartoon we really like and you, the reader can interpret as you see fit.
"There is no such thing as coincidence in this world — there is only inevitability"
-- Yuko Ichihara
If you take issue with any of the items on this page, then Contact the FDA, White House, and Congress and make your voice heard!

Comments and Discussion (Powered by the PricePlow Forum)